

アジレントの溶出試験器:溶出試験ワークフローの強化、簡略化、効率化を実現
アジレントは、製造されるほぼすべての剤形を試験するための溶出試験器を提供しています。R&D または QA/QC のいずれの場合においても、アジレントは、業界をリードする技術とソフトウェアにより、サンプルの分析を支援します。
![]()
![]()

in vitro 薬剤溶出試験では、標準化された条件下で薬剤が剤形(錠剤、カプセル)から溶液へ放出される速度を測定します。USP 総則 溶出試験では、溶出試験に必要な装置が規定されています。アジレントは、統一された薬局方要件に適合するほぼすべての剤形を試験できる、溶出試験器およびアクセサリを提供しています。アジレントの溶出試験器は、手動、半自動、またはオンライン分析モードで操作可能です。7 種類の溶出試験器があります。アジレントは、米国薬局方(USP)Apparatus 1(バスケット)、2(パドル)、3(レシプロケーティングシリンダ)、5(パドルオーバーディスク)、6(回転シリンダ)、7(レシプロケーティングディスク)を提供しています。

アジレントコミュニティでは、機器やメソッドについて質問したり、アドバイスを求めたりすることができます。専門的な情報を必要なタイミングで入手できます。
アジレントは、製造されるほぼすべての剤形を試験するための溶出試験器を提供しています。R&D または QA/QC のいずれの場合においても、アジレントは、業界をリードする技術とソフトウェアにより、サンプルの分析を支援します。

溶出試験液の容量としては、通常 500~900 mL を選択しますが、すべてのケースに適しているとは限りません。理想的には、溶出試験液の容量を選択する際は、薬剤の 3 倍以上を溶解できる容量にする必要があります。また、試験液の容量が多すぎて、その濃度での溶液の分析が困難にならないようにする必要もあります。
容量が 250 mL のベッセルの場合、パドルまたはバスケットをベッセルの内底から 25 mm の位置に設定する必要がありますか?サンプリングチューブの高さはどれくらいですか?溶出試験液の中間点からサンプリングする必要がありますか?1 L のベッセルで 500 mL の試験液を使用する場合はどうすればよいですか?サンプリングチップを中間点まで下げる必要がありますか?
高さは通常、ベッセルの底面から 25 mm の位置に設定します。USP に従い、試験液の表面とバスケットまたはパドルブレードの上部の中間点でサンプリングする必要があります。1 L のベッセルで 500 mL を使用する場合は、708-DS においてカニューラの高さが適切にキャリブレーションされていることを確認してください。設定するには、[Menu(メニュー)] > [Calibration(キャリブレーション)]を押します。『オペレータマニュアル』の 76 ページを参照してください。250 mL のベッセルの場合も、ベッセルの容量に応じて同じ方法で、同様に高さをキャリブレーションする必要があります。250 mL のベッセルを使用する際には、マニホールドアームの位置調整も必要になる場合があります。マニホールドの下面の各アームには、2 本の小さいネジが取り付けてあります。ネジを緩めてから、マニホールドアームを隣接する 2 つのネジ穴に移動させることができます。これにより、小型のベッセルではアームをシャフトに近づけ、1 L のベッセルでは遠ざけることが可能です。こうすることにより、適切に配置することができるため、カニューラが蒸発防止カバーやベッセル側面に接触して曲がることがなくなります。
経口フィルムは、取り扱いが困難な場合があります。以前、経口フィルムに対してバスケットが使用されているのを確認したことはありますが、より適切な解決策としては、Apparatus 5 ディスクを使用してフィルムをディスク上に配置するか、またはディスクを使用してフィルムを 2 枚のメッシュスクリーンの間に配置する方法があります。こうすることにより、フィルムを平坦に保持することができるため、放出がより均一になります。
シンク条件未満の試験液容量を用いた溶出試験メソッドを実施することは可能です。シンク条件は経験則であり、絶対値ではありません。溶解度が 3 倍ではなく 2 倍、2.5 倍などの場合でも、必ずしもメソッドが不適切であるとは限りません。シンク条件未満で作業する際には、メソッドの堅牢性を適切な状態に維持したままにするように注意する必要があります。シンク条件未満の場合には、わずかな変化に対してメソッドがより影響を受けやすくなる可能性があります。
溶出試験メソッドでは、良品と不良品の製剤を区別できる必要があります。理想的には、良品または不良品であることが分かっている溶出試験サンプルを用いた識別能試験を実施して、このメソッドを検証することが望ましいと考えられます。この検証が必要かどうかは、溶出の速さ、製品の意図など、いくつかの要因に依存します。これは、製品が安全かつ効果的であることを実証するために何が必要であるのかを判断するためのリスクベースの解析です。FDA やその他の規制機関は、この判断の基になる決定木を用意しています。
一般的に、2 つの溶出曲線や製品などをお互いに比較する場合は、n = 12 とします。比較には、5 点以上のタイムポイントが最適であり、85 % 以上となるタイムポイントは 1 点のみです。データセットの変動は、最初のタイムポイントでは < 20%、その他のタイムポイントではすべて < 10 % にする必要があります。
リソース:
はいオートサンプラの適格性評価では、装置が正常に動作することは保証されますが、これは、当該装置が現在のメソッドで使用するためにバリデーション済みであることは意味しません。メソッドは個別にバリデーションして、手動による結果と自動による結果の間に偏りがないことを示す必要があります。このバリデーションでは、クリーニングバリデーションやポンプパラメータの選択なども対象とする必要があります。
アプリケーションノート
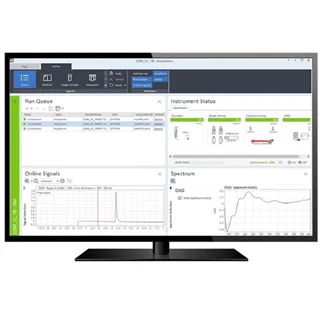









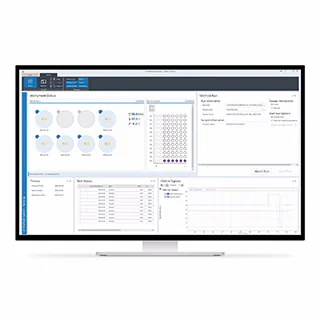
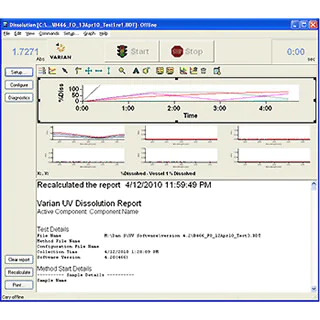
お問い合わせいただくか、または担当者からご連絡いたします。アプリケーション、教育イベント、ウェビナー、製品など、メソッドの最適化に役立つ最新情報の配信をご希望の方は、必ずご登録ください。




